EC Review Process
Do I need Ethics Approval?
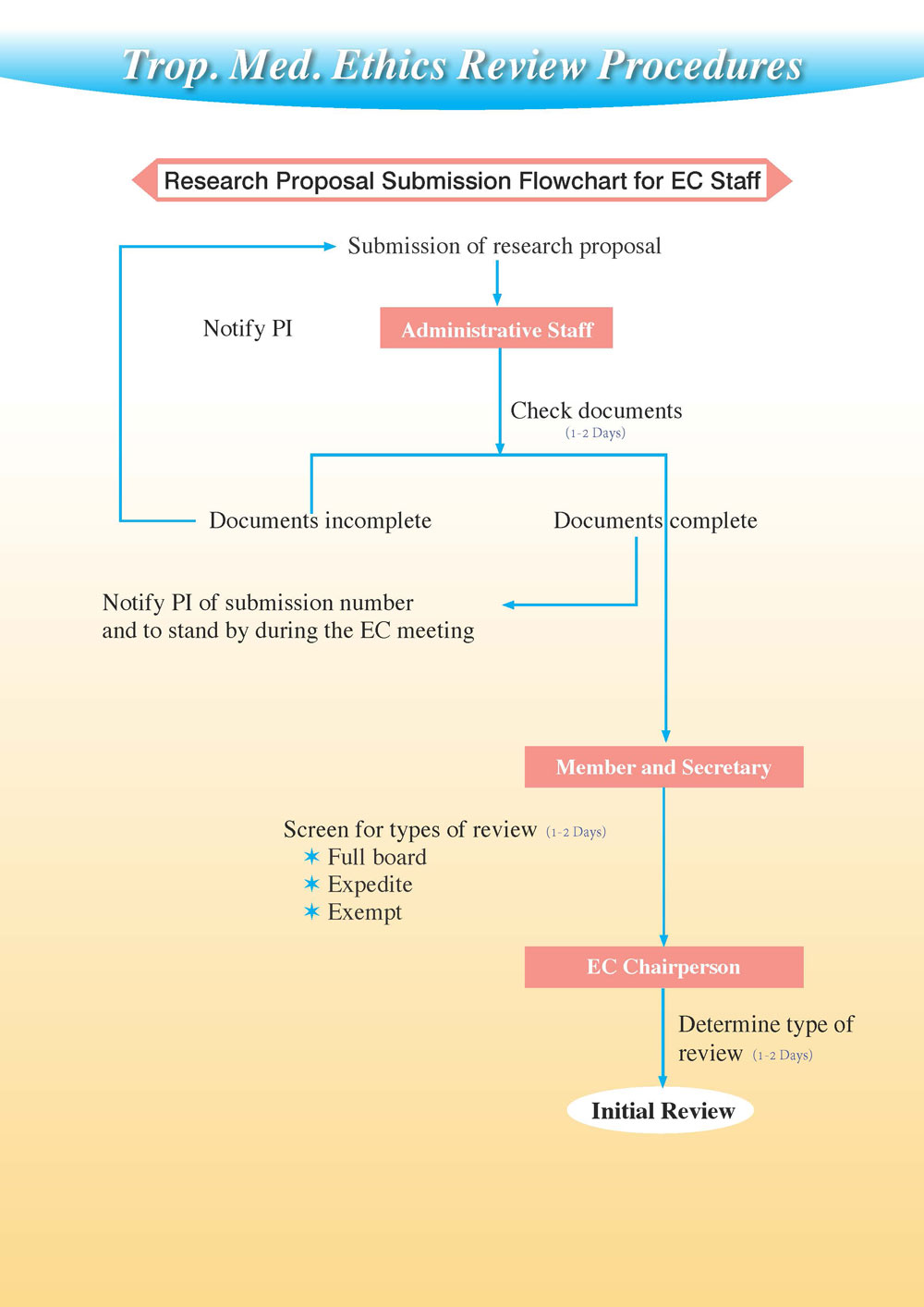
If your project involves human subjects, you require ethics committee approval. This is to ensure the safety of the subjects, protection of confidential information, and appropriate ethical treatment of those involved in your research. There are three routes to EC approval, full board review, expedited review, and exemption from review. To learn more about each route, please click the links below.
Process diagram
Full Board Review
Full Board Review is the standard review conducted by the EC. These reviews are conducted once a month. The schedule for the meetings can be found here
- Studies that cannot be reviewed and approved at an Exempt or Expedited review.
- Studies determined by the EC Chairperson as involving more than minimal risk.
- Studies involving elements, procedures or interventions that require additional provisions or safeguards, as stated by federal regulations and guidance.
- Studies involving highly vulnerable population, eg. HIV-infected persons, comatose patients, patients under critical care.
If you have further questions, please do not hesitate to contact the EC via email (tmectropmed@mahidol.ac.th) or visit our office on the 4th floor, of the 60th Anniversary of His Majesty the King's Accession to the Throne Building.
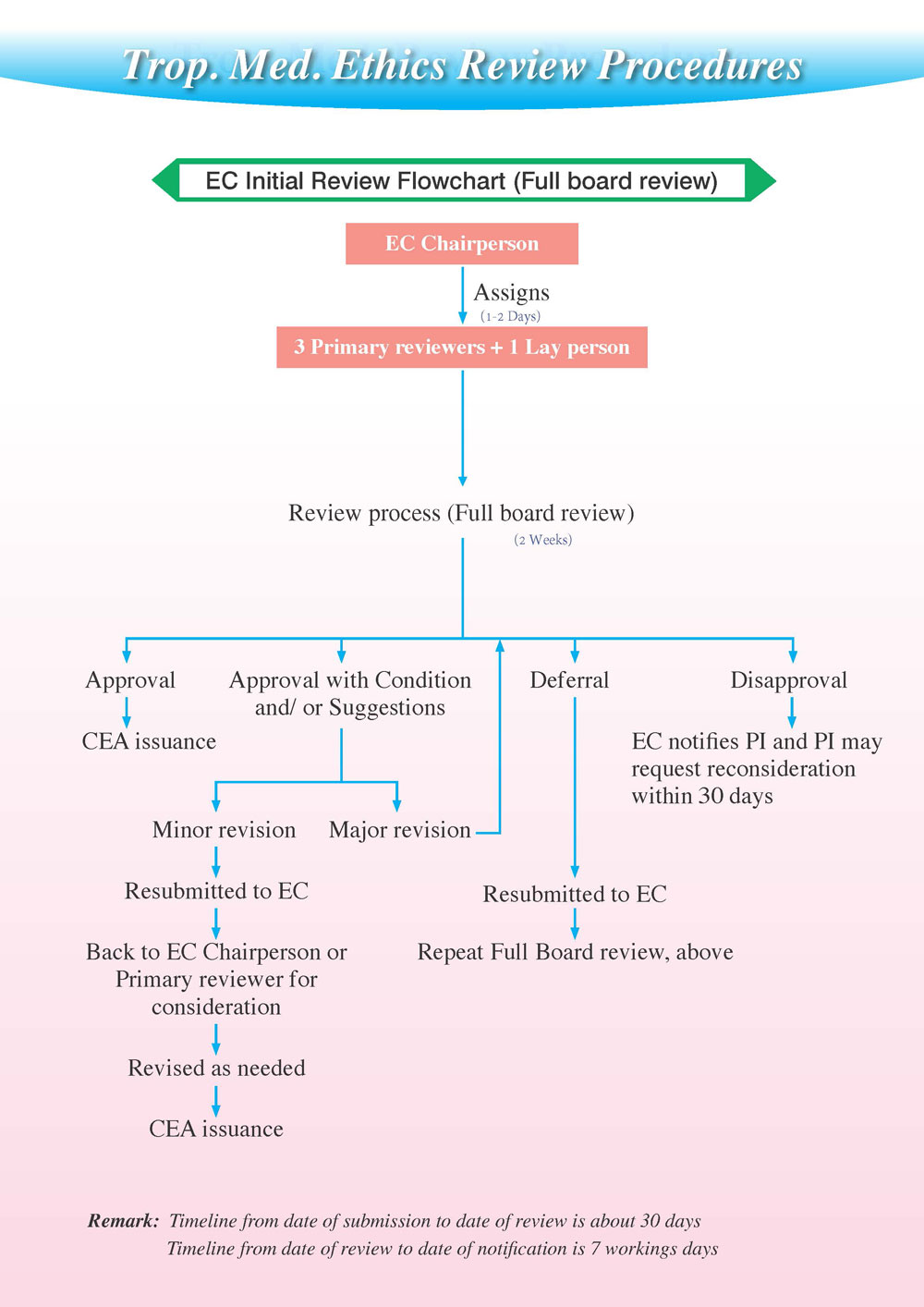
Once the research proposal and all appropriate submission forms have been submitted, the EC assistant secretary will distribute the materials to assigned reviewers seven working days before the scheduled meeting. The EC chairperson will assign three primary reviewers and one Lay member for each proposal. The assigned reviewers will complete a Reviewer's Assessment Form for Initial Review, while EC members who are not assigned reviewer of the review panel will read research proposal and be ready to submit their comments at the scheduled EC meeting. During the meeting, the following points must be discussed:
- Risks to the research participants are minimized.
- Risks to the research participants are reasonable in relationship to the anticipated benefits.
- Selection of the research participants is equitable.
- Informed consent will be obtained from the research participant or legally authorized representative.
- The proposal/protocol ensures research participant's safety through the monitoring of the data.
- The proposal/protocol ensures the research participant’s privacy and confidentiality of the data, if applicable.
- The investigator is appropriately qualified and has the facilities to ensure all aspects of the research will be conducted with regard for the safety and well-being of the research participants
After discussing the project according to the points above, members must reach a consensus to decide the outcome of each proposal. If there is no consensus, there will be a vote:
- Approved– approved as presented
- Modification prior to approval required (Major or Minor) – approved, subject to specific clarification/revision
- Defer – no decision can be made yet, pending evaluation of additional requested information
- Disapproved– the board has decided that they cannot ethically approve the research
If the EC decides on modification prior to approval required (Major or Minor), it must specify whether the changes will require a full board review or only primary reviewers.
Expedited Review
Expedited review is a speedier approval process available for certain projects which have specific characteristics:
Expedited review allows certain kinds of research to be reviewed and approved without convening a meeting of the EC. The EC will review certain categories of research through an expedited procedure only
Expedited review applies to research with the following characteristics
- Minimal risk
- May include identifiers (direct or indirect)
- Topics that are not sensitive OR may include some mildly sensitive topics, but where confidentiality is secure
- Populations may include vulnerable populations & others with adequate protection
- Consider a formal informed consent process OR justify a waiver of consent
- Requires continuing IRB review, at least annually
- Fits one of the 10 expedited categories, shown below
Categories of research which may be considered for expedited review include the following:
- Blood sample collection (routine medical checkup)
- Prospective collection of biological samples—noninvasive means
- Data collected though noninvasive means (routinely practiced in clinical settings)
- Materials (data, documents, specimens, etc.) have been collected or will be collected for non-research purposes
- Collection of voice, video or digital data for research purposes
- Low risk non-participatory observation, surveys, interviews, oral histories
- Research involving the collection or study of existing data, documents, medical records, stored, or identifiable leftover specimens, if these sources are available through authorized permission, or if the information is recorded by the investigator in such a manner that research participants cannot be identified directly or through identifiers linked to the research participants Leftover- or stored specimen can be stored as quality of specimen is available, but not more than ten (10) years. If the Principal Investigator wants to store the specimen for more than ten (10) years, the Principal Investigator must request permission from the EC Committee in writing.
- 8. Research proposal of multi-center study approved by the Central Research Ethics Committee (CREC) of which FTM EC is a member (See FTM ECS-009-RR), Research proposal for a multi-center study under a Memorandum of Understanding of Mahidol University has been considered by lead EC (where the FTM EC is the local EC), and project conducted outside Thailand by FTM staff/ student and has been approved by the local EC
- Continuing review of research previously approved by FTM EC as follows:
- where the research is permanently closed to the enrollment of new subjects; all subjects have completed all research-related interventions; and the research remains active only for long-term follow-up of subjects; or
- where no subjects have been enrolled and no additional risks have been identified; or
- where the remaining research activities are limited to data analysis
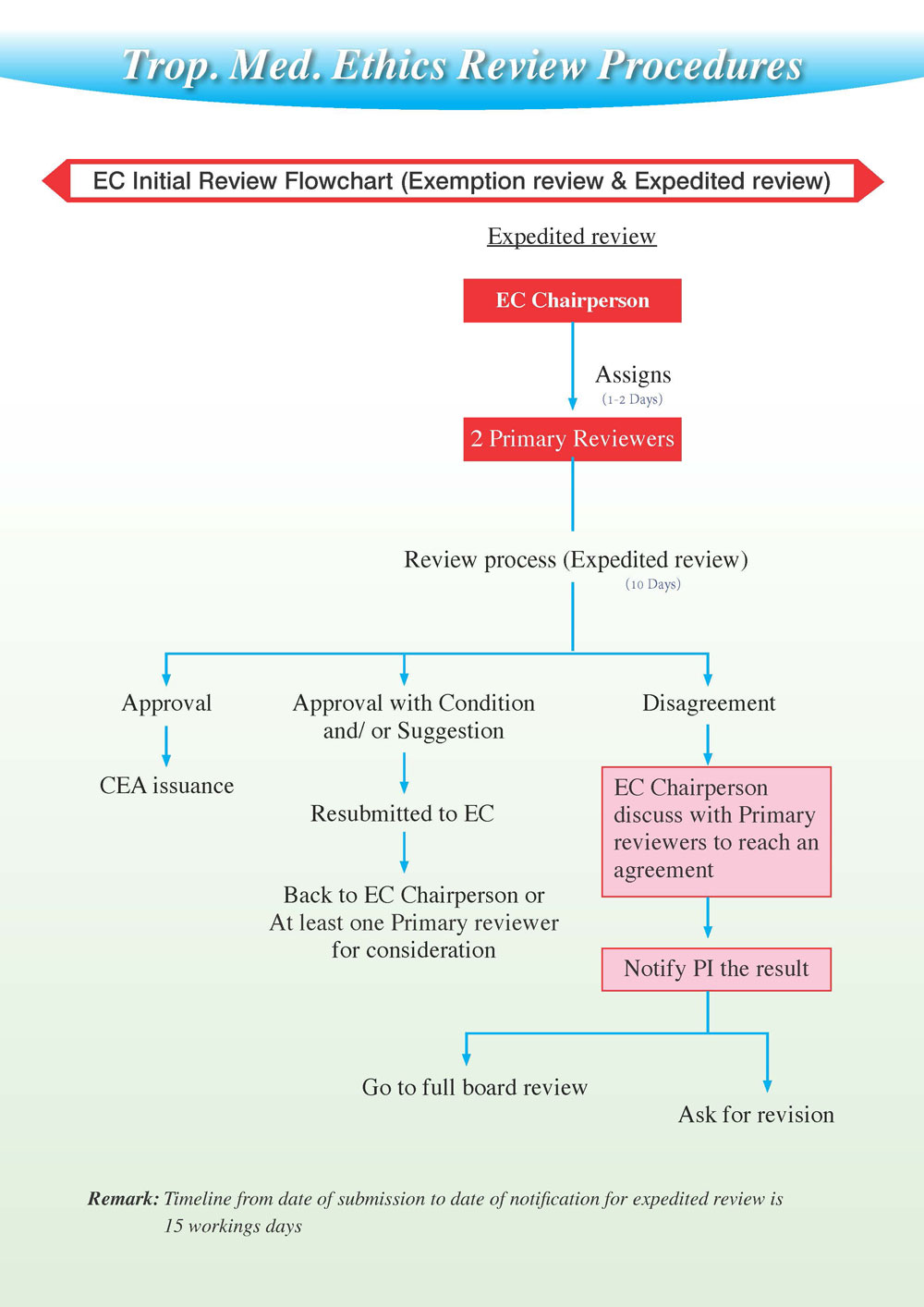
Exemption Review
Projects can be exempt from Ethics Committee review if they involve minimal risk to participants, do not include identifiers, or if the topic is deemed non-sensitive.
An exempt review applies to research that involves
- Minimal risk
- Exempt from continuing IRB review
- Fits one of 6 exempt categories below
Categories of research which may be considered for exemption include the following:
- Typical educational practices
- Educational tests, and surveys
- Research with elected public officials, appointed public officials, candidate for public office
- Existing data, documents, pathological specimens (if publicly available or rendered unidentifiable) and anonymous leftover specimens, data/ de-identified/ no identifiers maintained such as online survey
- Evaluation of public benefit service programs
- Taste and food quality evaluation and consumer acceptance studies

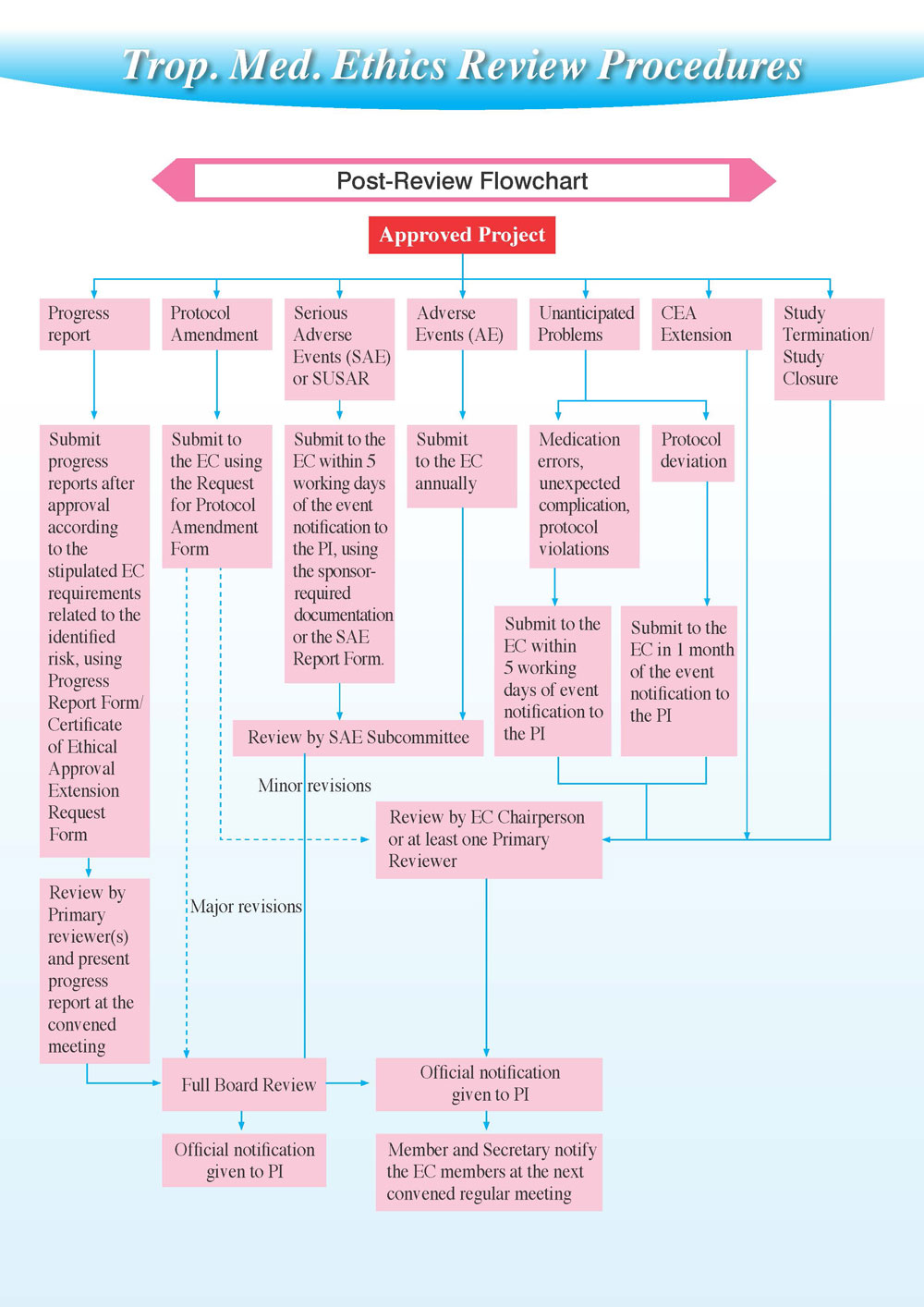
Post-Review
CEA is valid for one (1) year. Therefore, PI must extend the certificate using Progress Report Form/Certificate of Ethical Approval Extension Request Form (FTM ECF-008-RR) two (2) months prior to the expiry date if the study is not finished, and submit progress reports as stipulated by EC requirements related to the identified risk. In addition to these scheduled reports, PIs must submit reports in case of Serious Adverse Events (SAE), Adverse Events (AE), Serious Adverse Reactions (SUSARs), Protocol Amendments, or other unanticipated problems.
For reporting local SAE which are fatal or life threatening the PI must report to EC immediately, no later than twenty four (24) hours after the PI becomes aware of the event while for the local SAE which is non-fatal or non lifethreatening the PI must report to EC immediately, no later than seven (7) calendar days after the PI becomes aware of the event.
For reporting any non-local Serious Adverse Reactions sponsor must report non-local serious adverse reaction including SUSARs to EC at least every six (6) months accompanied by a brief report highlighting the main point of concern. Other adverse reactions that may increase risks to subjects, the sponsor must report to EC as soon as possible but no later than fifteen (15) calendar days. Other type of reports, the sponsor must report to EC at least every year or periodically or on request.
In case of the SAEs occurring in different countries of a multicenter project, the Investigator can report to the FTM EC in one (1) month of the event notification to PI.
For reporting local SUSARs which are fatal or life threatening sponsor must report to EC as soon as possible using CIOMS form, no later than seven (7) calendar days after the sponsor becomes aware of the event. If the initial report is incomplete, the sponsor must report to EC relevant follow-up information and complete report as soon as possible, within additional eight (8) calendar days. Sponsor must report any significant new information as a follow up report within fifteen (15) calendar days.
Local SUSARs which are non-fatal or non life threatening sponsor must report to EC as soon as possible using CIOMS form, no later than fifteen (15) calendar days after the sponsor becomes aware of the event. Further relevant follow-up information should be given as soon as possible.
The EC will require that PI report all Adverse Events related and not related to the study to the EC Chairperson. This must be accomplished in writing in one (1) year of the event notification to PI. The FTM EC will require that PI report all any unexpected situation affect serious to research project while the research participant is participating in a research study to the EC Chairperson (e.g., medication errors, unexpected complications, protocol violations). This must be accomplished in writing within five (5) working days of the event notification to PI, while the protocol deviations must be submitted in one (1) month.
These reports are reviewed by the EC Chairperson or by the primary reviewers (unless an amended protocol is considered to contain major revisions, in which case a full board review is necessary), while AE and SAE reports are reviewed by SAE Subcommittee, and once a decision has been made, the PI and the other EC members are notified.
- The following decisions can be reached in the post-review process:
- CEA extension - valid for another year
- Study closure - if the project has completed (all data collection and/or analysis has been finished), the final progress report results in a study closure.
- Study Termination - the study must be cancelled, either because circumstances prevent the PI to continue, or because the EC deems the project too high-risk to progress.
- Protocol Amendment - changes in project protocol can be accepted or denied only after review.
- SAE/Unexpected problems reports - These are submitted follow above timeline, and depending on the problems encountered, may result in a range of actions, from an acknowledgement without action, up to a CEA suspension/withdrawal. The protocol deviations must be submitted in one (1) month of the event notification to PI.